Geisel School of Medicine at Dartmouth, Memorial Sloan Kettering Cancer Center, Memorial Sloan Kettering Cancer Center
Background Gene expression signatures derived from RNA sequencing data have been associated with treatment outcomes for renal cell carcinoma (RCC) patients. Incorporating these RNA biomarkers into clinical practice is promising, yet its real-world applicability is heavily limited as RNA profiling is expensive, time-consuming, and requires specialized expertise for data analysis. In this study, we applied a deep neural network framework to identify the correlation between standard pathology images and underlying RNA signatures using hematoxylin and eosin (H&E) stained formalin-fixed paraffin-embedded (FFPE) whole slides of clear cell kidney tumors from The Cancer Genome Atlas (TCGA).
Methods We collected 496 H&E stained FFPE clear cell RCC whole-slide images and the RNA gene signatures for 496 patients from the TCGA database. We used this dataset to train and evaluate our weakly-supervised deep learning model. The model was iteratively trained using extracted patches from a slide and processed the patches through a convolutional neural network (CNN), pre-trained for the RCC subtypes classification task, to represent features. The features were aggregated and summarized to predict angiogenesis and myeloid infiltration scores. Performance was assessed by computing Pearson’s correlation coefficients.
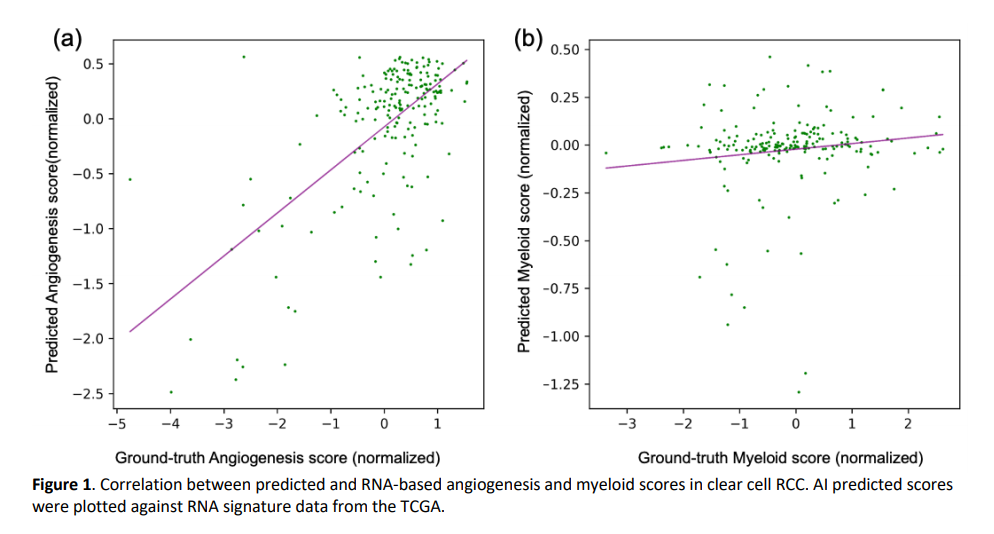
Results A separate group of 202 histology images was reserved as a test set. On this test set, the results of our weakly supervised method achieved a Pearson’s correlation of 0.65 (95% CI: 0.57-0.73) and 0.10 (95% CI: -0.04-0.23) with angiogenesis and myeloid scores from gold-standard RNA sequencing data, respectively (Figure 1).
Conclusions We proposed using deep learning-based AI techniques to process digitized histopathological images and estimate actionable signatures of angiogenesis from H&E stained slides. Our model showed promising results for predicting angiogenesis scores compared to myeloid scores. These results suggest the feasibility of this approach for estimating some digital biomarkers from H&E histopathology images and offering a rapid and cost-effective alternative to conventional RNA sequencing.