University of Michigan Ann Arbor, MI, United States
Grace Hile, Feiyang Ma, Amanda Victory, Bin Xu, Mehrnaz Gharaee-Kermani, Elisabeth Pedersen, Rachael Wasikowski, Celine Berthier, Vladimir Ognenovski, Allison Billi, Johann Gudjonsson and J. Michelle Kahlenberg, University of Michigan, Ann Arbor, MI
Background/Purpose: Skin disease in dermatomyositis (DM) is relapsing and often refractory to treatment, reflecting a lack of understanding of the mechanisms driving skin inflammation. DM rashes mimic cutaneous lupus erythematosus (CLE), and are frequently challenging to differentiate, particularly if disease is limited to the skin. Distinction is important, as systemic manifestations and prognosis between these diseases differ. Despite this, there has been limited research directly comparing DM and CLE. The objective of this study was to compare the immune cell composition and transcriptional changes in DM and CLE to advance our understanding of their distinct immunopathogenic mechanisms.
Methods: We used single-cell RNA sequencing (scRNA-seq) to investigate cellular transcriptomes in normal appearing ‘non-lesional’ and lesional skin samples from 8 patients with DM and 8 patients with CLE; all patients met ACR/EULAR criteria for DM and SLE, respectively. Peripheral blood mononuclear cells (PBMCs) were also collected from 5 of 8 DM and all CLE patients. Disease groups were paired with 8 sex-matched healthy control (HC) skin biopsies and healthy control PBMC samples. Skin biopsies were processed into single-cell suspensions. Libraries were generated from skin cells and PBMCs via the 10X Chromium system and sequenced via Illumina NovaSeq 6000. Samples were merged into a single expression matrix and analyzed using Seurat. Enrichr pathway analysis was used to identify pathways differentiating DM from CLE.
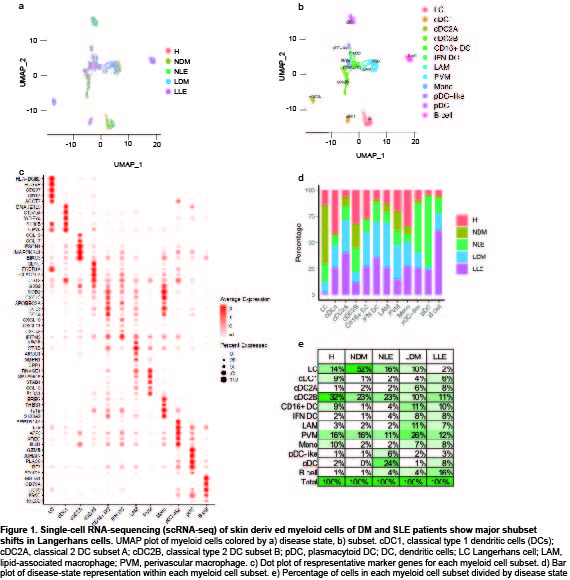
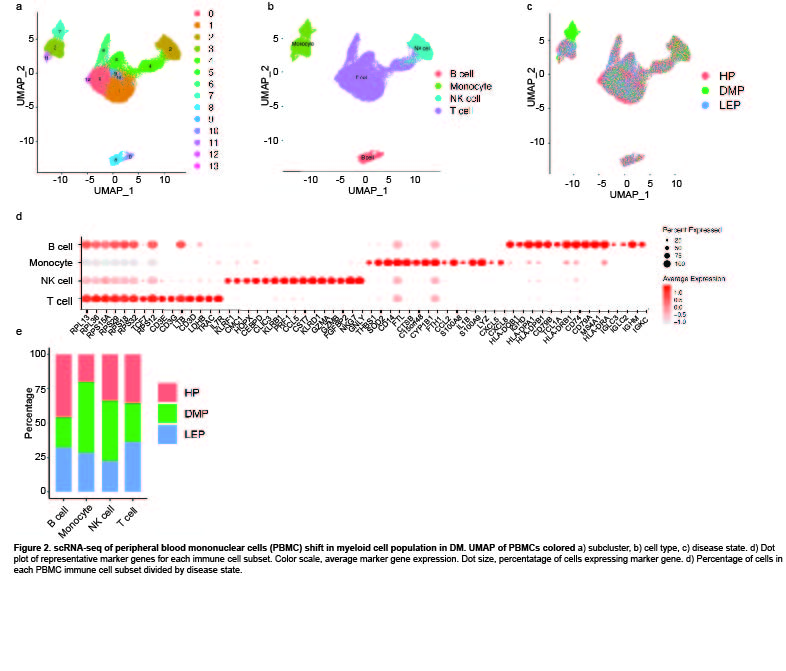
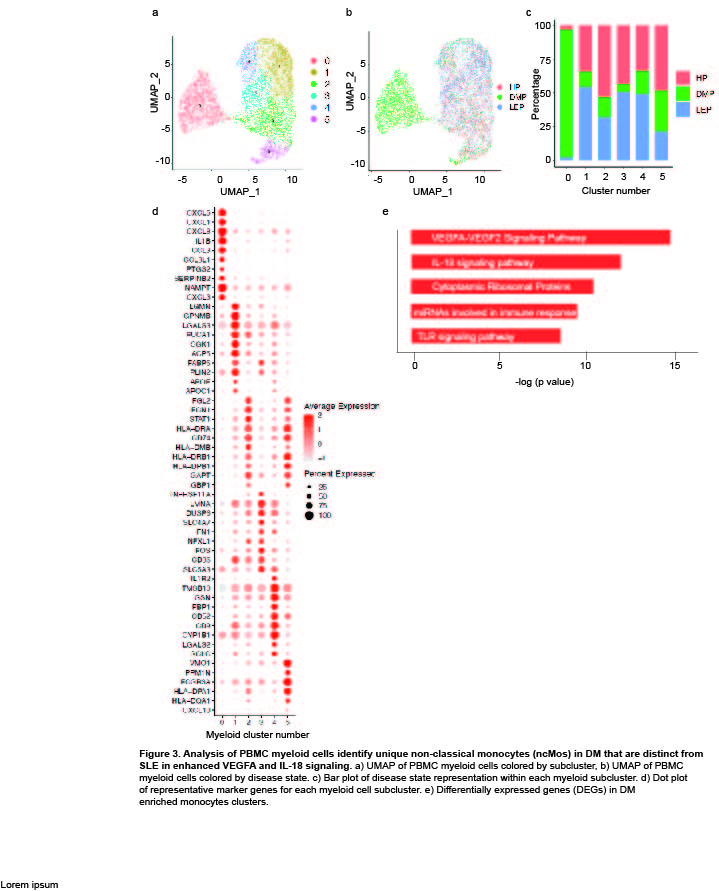
Results: Clustering and annotation of myeloid cells from skin biopsies revealed 11 myeloid cell subsets, along with B cells, with largely distinct marker genes (Figure 1a-c). Myeloid cell subsets showed variability in representation among disease states. Among the most striking differences was that Langerhans cells (LC; CD207, CD1A) were dramatically expanded in non-lesional DM samples (Fig 1d), constituting 52% of all myeloid cells in non-lesional samples versus only 14% in HC (Fig 1e). Conversely, lesional LE skin showed contraction of LCs, representing only 2% of myeloid cells in those samples. Examination of PBMCs by scRNA-seq (Fig 2) revealed expansion of monocytes in DM patient PBMCs (Fig 2e), and several DM patients showed a disease-specific, transcriptionally distinct monocyte subset (Fig 2c). Monocyte sub-clustering and examination of marker genes revealed subclusters with vast majority derived from DM PBMCs (Fig 3). Top canonical pathways enriched in DM-enriched subclusters versus LE related to VEGF signaling pathway (p=10-14), IL-18 signaling pathway (p=10-13) and cytoplasmic ribosomal proteins (p=10-9), all relevant pathways in DM, which is characterized by vasculopathy and elevated IL-18 expression in the skin and muscle.
Conclusion: Our data outline the composition of myeloid cell populations in DM skin and PBMCs compared to SLE. We demonstrate increases in LC in DM skin compared to SLE and a strikingly DM-enriched monocyte population that differentiates from CLE by enriched VEGF2A and IL-18 signaling. Further investigation into the pathogenic role of these monocytes is needed to identify their contribution to DM pathogenesis.
Disclosures: G. Hile, None; F. Ma, None; A. Victory, None; B. Xu, None; M. Gharaee-Kermani, None; E. Pedersen, None; R. Wasikowski, None; C. Berthier, None; V. Ognenovski, None; A. Billi, None; J. Gudjonsson, None; J. Kahlenberg, Q32 Bio, Celgene/Bristol Myers Squibb, Ventus Therapeutics, Rome Therapeutics, Janssen, AstraZeneca, Eli Lilly, GlaxoSmithKline, Bristol Myers Squibb, Avion Pharmaceuticals, Provention Bio, Aurinia Pharmaceuticals, Boehringer Ingelheim.