
Poster Session D
Immunobiology
Vinh Dang, BS
Children's Hospital of Philadelphia
Philadelphia, PA, United States
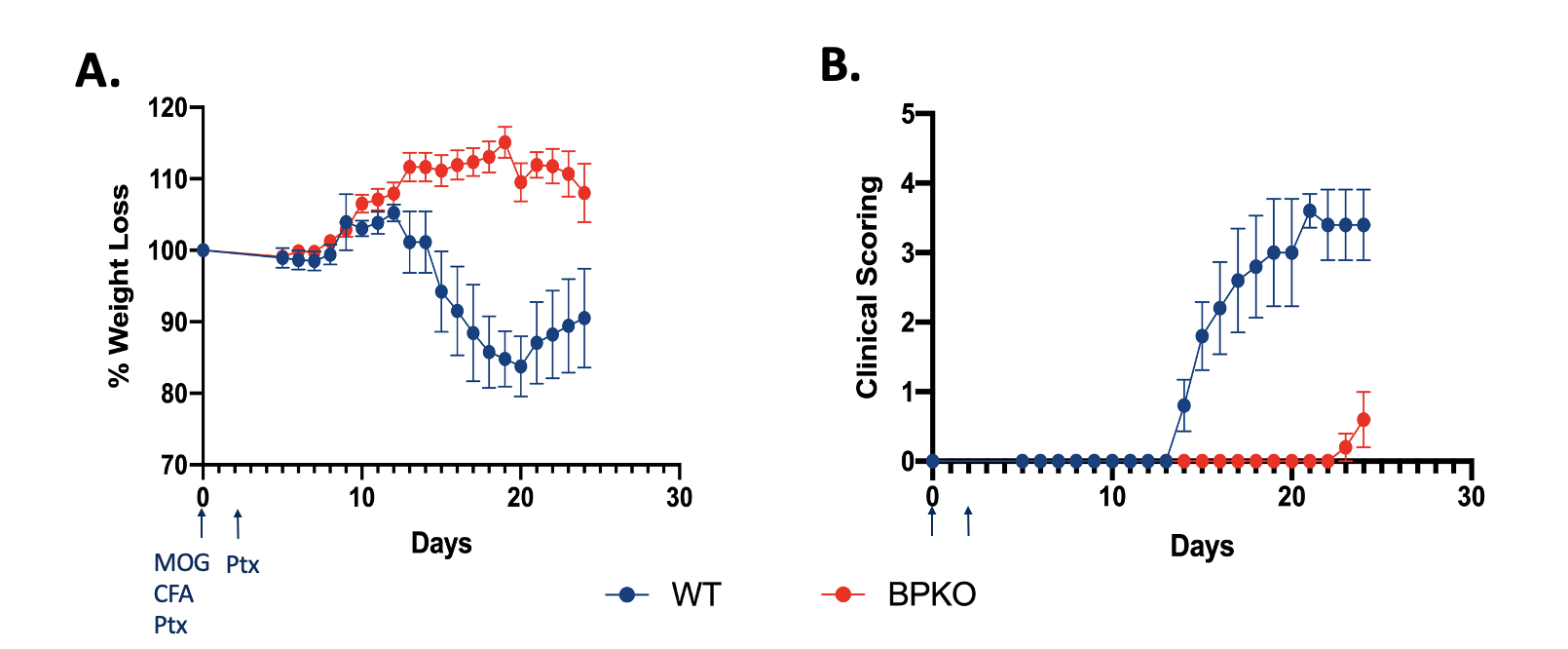 Unopposed IL-18 protects mice from experiencing clinical outcomes of EAE 24 days post immunization with MOG peptide and Pertussis Toxin. (A) Percent weight loss of Il18bp-/- mice in comparison to WT mice. (n=5) (B) Clinical outcomes of Il18bp-/- mice compared to WT mice. (n=5)
Unopposed IL-18 protects mice from experiencing clinical outcomes of EAE 24 days post immunization with MOG peptide and Pertussis Toxin. (A) Percent weight loss of Il18bp-/- mice in comparison to WT mice. (n=5) (B) Clinical outcomes of Il18bp-/- mice compared to WT mice. (n=5)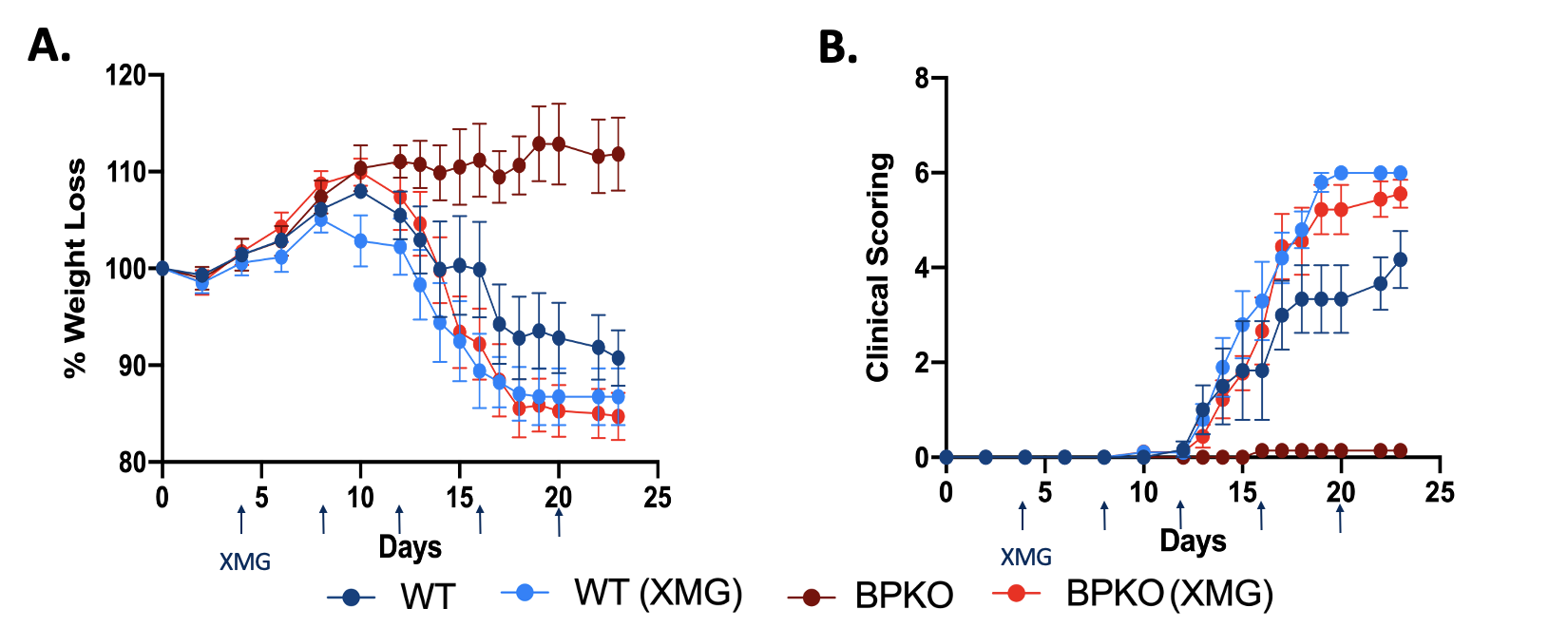 Blocking IFNg in Il18bp-/- mice renders mice susceptible to EAE meaning the protection offered by unopposed IL-18 is IFNg dependent. (A) Percent weight loss of Il-18bpKO mice in comparison to WT mice untreated and treated with XMG 1.2. (n=5) (B) Clinical outcomes of Il18bp-/- mice compared to WT mice untreated and treated with XMG 1.2. (n=5)
Blocking IFNg in Il18bp-/- mice renders mice susceptible to EAE meaning the protection offered by unopposed IL-18 is IFNg dependent. (A) Percent weight loss of Il-18bpKO mice in comparison to WT mice untreated and treated with XMG 1.2. (n=5) (B) Clinical outcomes of Il18bp-/- mice compared to WT mice untreated and treated with XMG 1.2. (n=5)